马上注册,结交更多好友,享用更多功能,让你轻松玩转社区。
您需要 登录 才可以下载或查看,没有账号?立即注册

x
《每日科学》(2012年9月6日)——2012年在芝加哥胸腔肿瘤多学科研讨会上发表的研究结果表明,单克隆抗体bavituximab结合多西他赛的化疗可使总体应答率增加一倍,并且提高已接受早期化疗治疗方案的晚期非鳞状非小细胞肺癌(NS-NSCLC)患者的无进展生存期和总体生存期。
本次研讨会由美国临床肿瘤协会(ASCO)、美国放射肿瘤协会(ASTRO)、国际肺癌研究协会(IASLC)和芝加哥大学赞助。
多西他赛是标准的IIIB期和IV期的非鳞状非小细胞肺癌(NS-NSCLC)患者的二线治疗药物。研究人员在以多西他赛加安慰剂作为对照组的II期、9中心、双盲、随机临床实验中评估了多西他赛加1毫克或3毫克bavituximab的有效性和安全性。
1mg bavituximab实验组的总体应答率为15%,3mg的为17.9%,约为对照组总体应答率7.9%的两倍。Bavituximab组的中位无进展生存期为4.2个月和4.5个月,而对照组为3个月。
该临床实验在18个月后解盲,此时,对照组的中位生存期达到5.4个月,61%的患者死亡;然而,不管bavituximab组是否达到该中位生存期,但其仅有不到35%的患者死亡。
“此次严谨的II期临床试验表明,对晚期NSCLC患者二线化疗来说,bavituximab结合多西他赛给药不仅耐受性良好,而且可以提高应答率、无进展生存期和总生存期,”David Gerber,一位医师,该临床实验的主要研究者,并在在达拉斯得克萨斯大学西南医学中心专门从事肺癌治疗的内科助理教授如此说。“如果III期临床试验证实这些研究结论,bavituximab可能将成为该极具挑战性疾病的标准治疗方案的重要组成部分。” |
|
|
个人公众号:treeofhope
|
|
|
|
共5条精彩回复,最后回复于 2014-1-16 16:13

尚未签到

尚未签到

尚未签到

尚未签到
美国生物制药公司Peregrine旗下用于二线非小细胞肺癌(NSCLC)治疗的重要试验性免疫治疗药物巴维昔单抗(Bavituximab)获得美国食品药品管理局(FDA)快速通道审评资格。
该公司最近启动了一项名为SUNRISE的关键3期临床试验,该试验将巴维昔单抗+多西他赛与安慰剂+多西他赛用于非小细胞肺癌治疗进行了对比。SUNRISE是一项全球、随机、双盲、安慰剂对照3期临床试验,该试验旨在评价巴维昔单抗+多西他赛用于二线非小细胞肺癌患者的安全性、耐受性和有效性。
Peregrine制药法规事务主管Robert Garnick说,“快速通道审评资格对SUNRISE临床试验项目来说是个里程碑,代表巴维昔单抗朝着上市迈进了一步。IIIb/IV期临床试验受试者为600多名经过标准一线治疗后病情仍有进展的非鳞、NSCLC患者,全球100多家临床单位参与了这项临床试验项目。
在临床试验中,所有患者将进行6个周期,每个周期为21天的多西他赛(75 mg/m2)加每周一次巴维昔单抗或安慰剂输液治疗,直到病情出现进展或毒性反应。根据该公司的说法,巴维昔单抗是一种新类型的以磷脂酰丝氨酸为靶点的单克隆抗体,是治疗癌症的一种新途径。 |
|
|
|
|
|
|
尚未签到
美国FDA有三条特别审批通道,即快速通道(Fast Track)、优先审评(Priority Review)、加速批准(Accelerated Approval)。进入特别通道的条件有两个,即目前无有效药物(no current therapy),新药能填补空白(first available treatment),或者新药在有效性或安全性上有明显优势(have advantages over existing treatments)。
快速通道(Fast Track)
FDA1988年引入快速通道,由制药企业主动申请(可以在药物研发的任何阶段),FDA在收到申请后60天内给出答复。对进入快速通道的药物,FDA将进行早期介入,就哪些试验该做哪些试验可以不做等内容提出指导意见,以达到让该产品在研发过程中少走弯路,加快整个研发过程的效果。另外,药企还可分阶段递交申报资料,而不需要一次性递交全部材料才进行审评。
加速批准(Accelerated Approval)
药物临床试验拿到clinical outcome需要很长时间,在1992年FDA引入加速批准通道,用surrogate endpoint代替clinical endpoint,先批准后验证,如果上市后验证了临床疗效,则FDA维持原先的批准。例如抗癌药的clinical endpoint是增加患者的生存率或延长存活时间,但直接拿到这样的临床结论需要很长的时间,这时可以采用surrogate endpoint替代,即研究肿瘤的萎缩程度。如果药物能缩小肿瘤,那么基本可以认定它能延长患者存活时间。
优先审评(Priority Review)
1992年FDA引入优先审评,优先审评不仅针对严重疾病(serious diseases),也适用于普通疾病(less serious illnesses),能否进入优先审评的关键在于是否有优于现有治疗手段的潜力(have the potential to provide significant advances in treatment)。优先审评需药企在提交NDA或BLA时主动申请,FDA将在45天内给出答复。与快速通道、加速批准不同的是,优先审评只针对审评阶段,而不加速临床试验。根据Prescription Drug User Act,快速审评的周期为6个月,而标准审评(Standard Review)周期为10个月。
Reference: http://www.fda.gov/forconsumers/ ... apies/ucm128291.htm |
|
|
个人公众号:treeofhope
|
|
|
|
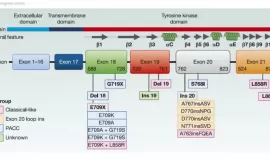
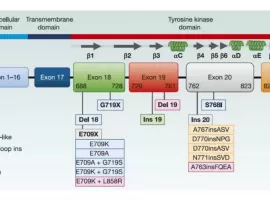 EGFR罕见突变种类多,哪种靶向药治疗
作者:seacat
针对EGFR罕见突变(排除外显子20插入 Exon 20ins),目前获批的靶向药是
EGFR罕见突变种类多,哪种靶向药治疗
作者:seacat
针对EGFR罕见突变(排除外显子20插入 Exon 20ins),目前获批的靶向药是
 大病恢复,野生灵芝确证有效
分享一个良方,希望能帮助到有需要的人。姑妈和大舅妈都生过重病(这里不说是啥病了哈
大病恢复,野生灵芝确证有效
分享一个良方,希望能帮助到有需要的人。姑妈和大舅妈都生过重病(这里不说是啥病了哈
 ADC药物如何狙击肺癌——2024.8.27董
ADC药物如何狙击肺癌——2024.8.27董晓荣教授直播笔记
直播链接:
https://live.yuaig
ADC药物如何狙击肺癌——2024.8.27董
ADC药物如何狙击肺癌——2024.8.27董晓荣教授直播笔记
直播链接:
https://live.yuaig
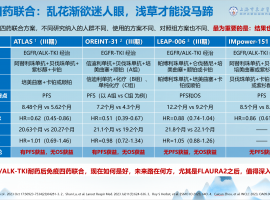 EGFR突变肺癌治疗策略及进展研究——
EGFR突变肺癌治疗策略及进展研究——周斐教授讲座学习笔记
直播链接:
https://live.
EGFR突变肺癌治疗策略及进展研究——
EGFR突变肺癌治疗策略及进展研究——周斐教授讲座学习笔记
直播链接:
https://live.
 求助 K药和替雷利珠如何选择
父亲9月刚查出非小细胞低分化癌伴随左肾上腺和第10胸椎转移PDL1高表达(TPS=90%) SMARC
求助 K药和替雷利珠如何选择
父亲9月刚查出非小细胞低分化癌伴随左肾上腺和第10胸椎转移PDL1高表达(TPS=90%) SMARC


 提升卡
提升卡 置顶卡
置顶卡 沉默卡
沉默卡 喧嚣卡
喧嚣卡 变色卡
变色卡 千斤顶
千斤顶 显身卡
显身卡